
Macayla Grace Smoak - May 22, 2001 - May 22, 2010
Where It All Started...In April 2005, Macayla started having episodes that lasted a couple of seconds each, and they looked like they could seizures. She fell a couple of times for seemingly no reason. We had a CT scan, and a couple of days later, she started running a fever. This exacerbated the collapsing. It was as if all the bones were suddenly removed from her body, causing her to just go limp.
|

|
This led to admission to the hospital, where we spent a few days running tests with no helpful results. Her MRI was declared to be normal, and her condition was blamed on viral meningitis. After release from the hospital, her episodes continued, and she started falling often. She reverted to crawling because she fell so often. She even began to anticipate her falls. She would look down at her body and whimper just before she collapsed.
We had a 72-hour EEG to check for seizures, and we discovered that she was having absence seizures, twice as many as we could observe outwardly. We put her on medicine, Lamictal, and it took several weeks to build up to a therapeutic dose.
The medicine, however, never seemed to keep up with the seizure activity. We sought a second opinion and discovered that Macayla's first MRI was not normal as previously thought. There was slight under development in her cerebellum, but due to the fact we only had one MRI, we did not know if she had been born this way or not. Her seizures were not typical, and the doctor called it myoclonic astatic epilepsy. She was having absence seizures with myoclonic and atonic components. This meant here seizures could be undetectable or have various, unpredictable components. She may simply blink repeatedly, lose tone in part or all of her body, or have jerking in part or all of her body. The seizures typically lasted a few seconds at first, and she was having 100 seizures per day. Our doctor warned us that it would take quite a bit of trial and error to get the right combination of medicines to control the seizures, if at all. We never got full control of them. Epilepsy is a symptom of something else. According to our doctor, half of the time that something else is never discovered.
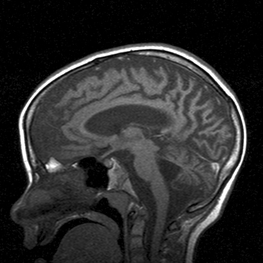
We went through what one doctor called "The Million-Dollar Workup" to try to find the cause for Macayla's epilepsy, but to no avail. In the meantime, Macayla continued to digress developmentally. The medicines did not control her seizures. By November, we knew something was going terribly wrong, but we just could not get a diagnosis. We had a second MRI in December 2005, and the image showed her cerebellum looked worse than it did in April. This confirmed that the "under development" in her cerebellum was actually atrophy. Further, the rest of the brain was beginning to show atrophy as well.
This confirmed what we already suspected. We were facing a progressive, degenerative brain disorder. Our doctor tested for Battens Disease, and the positive result came back the week after Christmas. The progression of Battens is unpredictable and unique to each person. The major symptoms are blindness, seizures, and developmental digression. There is no cure or effective treatments for Battens Disease.
We had a 72-hour EEG to check for seizures, and we discovered that she was having absence seizures, twice as many as we could observe outwardly. We put her on medicine, Lamictal, and it took several weeks to build up to a therapeutic dose.
The medicine, however, never seemed to keep up with the seizure activity. We sought a second opinion and discovered that Macayla's first MRI was not normal as previously thought. There was slight under development in her cerebellum, but due to the fact we only had one MRI, we did not know if she had been born this way or not. Her seizures were not typical, and the doctor called it myoclonic astatic epilepsy. She was having absence seizures with myoclonic and atonic components. This meant here seizures could be undetectable or have various, unpredictable components. She may simply blink repeatedly, lose tone in part or all of her body, or have jerking in part or all of her body. The seizures typically lasted a few seconds at first, and she was having 100 seizures per day. Our doctor warned us that it would take quite a bit of trial and error to get the right combination of medicines to control the seizures, if at all. We never got full control of them. Epilepsy is a symptom of something else. According to our doctor, half of the time that something else is never discovered.
We went through what one doctor called "The Million-Dollar Workup" to try to find the cause for Macayla's epilepsy, but to no avail. In the meantime, Macayla continued to digress developmentally. The medicines did not control her seizures. By November, we knew something was going terribly wrong, but we just could not get a diagnosis. We had a second MRI in December 2005, and the image showed her cerebellum looked worse than it did in April. This confirmed that the "under development" in her cerebellum was actually atrophy. Further, the rest of the brain was beginning to show atrophy as well.
This confirmed what we already suspected. We were facing a progressive, degenerative brain disorder. Our doctor tested for Battens Disease, and the positive result came back the week after Christmas. The progression of Battens is unpredictable and unique to each person. The major symptoms are blindness, seizures, and developmental digression. There is no cure or effective treatments for Battens Disease.
|
|
Over time, affected children suffer mental impairment, worsening seizures, and progressive loss of sight and motor skills. Eventually, children with Batten Disease/NCL become blind, bedridden, and unable to communicate. Presently, Battens is always fatal. Batten Disease is not contagious or, at this time, preventable.
Types of Battens:
There are four main types of NCL, including two forms that begin earlier in childhood and a very rare form that strikes adults. The symptoms are similar, but they become apparent at different ages and progress at different rates.
Infantile NCL (Santavuori-Haltia disease): begins between about 6 months and 2 years of age and progresses rapidly. Affected children fail to thrive and have abnormally small heads (microcephaly). Also typical are short, sharp muscle contractions called myoclonic jerks. Initial signs of this disorder include delayed psychomotor development with progressive deterioration, other motor disorders, or seizures. The infantile form has the most rapid progression and children live into their mid childhood years.
Late Infantile NCL (Jansky-Bielschowsky disease): begins between ages 2 and 4. The typical early signs are loss of muscle coordination (ataxia) and seizures along with progressive mental deterioration. This form progresses rapidly and ends in death between ages 8 and 12. Macayla had this form of Battens.
Juvenile NCL (Batten Disease): begins between the ages of 5 and 8 years of age. The typical early signs are progressive vision loss, seizures, ataxia or clumsiness. This form progresses less rapidly and ends in death in the late teens or early 20s, although some may live into their 30s.
Adult NCL (Kufs Disease or Parry's Disease): generally begins before the age of 40, causes milder symptoms that progress slowly, and does not cause blindness. Although age of death is variable among affected individuals, this form does shorten life expectancy.
There are four additional diseases included in the Batten Disease/NCL group:
Finnish Late Infantile - identified in Finland.
Variant Late Infantile - identified in Costa Rica, South America, Portugal and other nations.
Turkish Late Infantile - identified in Turkey.
Northern Epilepsy/ERMP - Epilepsy with Mental Retardation - identified in Finland.
How Many People are Affected:
Batten Disease/NCL is relatively rare, occurring in an estimated 2 to 4 of every 100,000 births in the United States. The diseases have been identified worldwide. Although NCLs are classified as rare diseases, they often strike more than one person in families that carry the defective gene.
How Is Battens Passed On:
Childhood NCLs are autosomal recessive disorders; that is, they occur only when a child inherits two copies of the defective gene, one from each parent. When both parents carry one defective gene, each of their children faces one-in-four chance of developing NCL. At the same time, each child also faces a one-in-two chance of inheriting just one copy of the defective gene. Individuals who have only one defective gene are known as carriers, meaning they do not develop the disease, but they can pass the gene on to their own children.
Adult NCL may be inherited as an autosomal recessive (Kufs) or, less often, as an autosomal dominant (Parrys) disorder . In autosomal dominant inheritance, all people who inherit a single copy of the disease gene develop the disease. As a result, there are no unaffected carriers of the gene.
Types of Battens:
There are four main types of NCL, including two forms that begin earlier in childhood and a very rare form that strikes adults. The symptoms are similar, but they become apparent at different ages and progress at different rates.
Infantile NCL (Santavuori-Haltia disease): begins between about 6 months and 2 years of age and progresses rapidly. Affected children fail to thrive and have abnormally small heads (microcephaly). Also typical are short, sharp muscle contractions called myoclonic jerks. Initial signs of this disorder include delayed psychomotor development with progressive deterioration, other motor disorders, or seizures. The infantile form has the most rapid progression and children live into their mid childhood years.
Late Infantile NCL (Jansky-Bielschowsky disease): begins between ages 2 and 4. The typical early signs are loss of muscle coordination (ataxia) and seizures along with progressive mental deterioration. This form progresses rapidly and ends in death between ages 8 and 12. Macayla had this form of Battens.
Juvenile NCL (Batten Disease): begins between the ages of 5 and 8 years of age. The typical early signs are progressive vision loss, seizures, ataxia or clumsiness. This form progresses less rapidly and ends in death in the late teens or early 20s, although some may live into their 30s.
Adult NCL (Kufs Disease or Parry's Disease): generally begins before the age of 40, causes milder symptoms that progress slowly, and does not cause blindness. Although age of death is variable among affected individuals, this form does shorten life expectancy.
There are four additional diseases included in the Batten Disease/NCL group:
Finnish Late Infantile - identified in Finland.
Variant Late Infantile - identified in Costa Rica, South America, Portugal and other nations.
Turkish Late Infantile - identified in Turkey.
Northern Epilepsy/ERMP - Epilepsy with Mental Retardation - identified in Finland.
How Many People are Affected:
Batten Disease/NCL is relatively rare, occurring in an estimated 2 to 4 of every 100,000 births in the United States. The diseases have been identified worldwide. Although NCLs are classified as rare diseases, they often strike more than one person in families that carry the defective gene.
How Is Battens Passed On:
Childhood NCLs are autosomal recessive disorders; that is, they occur only when a child inherits two copies of the defective gene, one from each parent. When both parents carry one defective gene, each of their children faces one-in-four chance of developing NCL. At the same time, each child also faces a one-in-two chance of inheriting just one copy of the defective gene. Individuals who have only one defective gene are known as carriers, meaning they do not develop the disease, but they can pass the gene on to their own children.
Adult NCL may be inherited as an autosomal recessive (Kufs) or, less often, as an autosomal dominant (Parrys) disorder . In autosomal dominant inheritance, all people who inherit a single copy of the disease gene develop the disease. As a result, there are no unaffected carriers of the gene.
